Long QT Syndrome: Difference between revisions
No edit summary |
|||
| (14 intermediate revisions by 5 users not shown) | |||
| Line 5: | Line 5: | ||
|moderator= [[user:Pgpostema|P.G. Postema, MD]] | |moderator= [[user:Pgpostema|P.G. Postema, MD]] | ||
|editor= | |editor= | ||
}} | }} | ||
The '''Long QT Syndrome (LQTS)''' is characterized on the ECG by prolongation of the [[Conduction#The_QT_interval|heart rate corrected QT interval]]. This was first recognized by Dr. Jervell and Dr. Lange-Nielsen in 1957. They described 4 children with a long QT interval which was accompanied by hearing deficits, sudden cardiac death and an autosomal recessive inheritance.<cite>Lang1957</cite> | |||
The '''Long QT Syndrome (LQTS)''' is characterized on the ECG by prolongation of the [[Conduction#The_QT_interval|heart rate corrected QT interval]]. This was first recognized by | |||
The Long QT syndrome may be divided into two distinct forms: congenital Long QT syndrome and acquired Long QT syndrome. These forms may however overlap when QT prolongation due to medication occurs in a patient with congenital Long QT syndrome. | |||
<gallery> | |||
File:acquired_longQT.jpg|A 12-lead ECG of a patient with acquired long QT syndrome. Notice the QT prolongation. The QTc is about 640ms. | |||
File:Lqts1.png|A 12 lead ECG of a patient with genetically proven LQTS1 | |||
File:Lqts2.png|A 12 lead ECG of a patient with genetically proven LQTS2 | |||
File:Lqts3.png|A 12 lead ECG of a patient with genetically proven LQTS3 | |||
</gallery> | |||
===Diagnosis=== | ===Diagnosis=== | ||
*The diagnosis is by | *The diagnosis is by measurement of the [[Conduction#The_QT_interval|heart rate-corrected QT interval]] on the ECG, which can be calculated with the [[QTc calculator]]. | ||
* | *Sometimes the QT interval can be difficult to assess. Read the [[Difficult_QT|guidelines for measurement of difficult QT interval]]. | ||
*A QTc of > 500ms in patients with Long QT Syndrome is associated with an increased risk for sudden death.<cite>Priori</cite> | *A QTc of > 500ms in patients with Long QT Syndrome is associated with an increased risk for sudden death.<cite>Priori</cite> | ||
*In patients suspected of LQTS (e.g. family members of known LQTS patients) a QTc > 430ms makes it likely that a LQTS gene defect is present.<cite>Hofman</cite> | *In patients suspected of LQTS (e.g. family members of known LQTS patients) a QTc > 430ms makes it likely that a LQTS gene defect is present.<cite>Hofman</cite> | ||
| Line 104: | Line 108: | ||
| '''Trigger''' | | '''Trigger''' | ||
| '''Eponyme''' | | '''Eponyme''' | ||
| [[w:OMIM|OMIM™]] link | | '''[[w:OMIM|OMIM™]] link''' | ||
|- | |- | ||
! LQTS1 | ! LQTS1 | ||
| Line 146: | Line 150: | ||
|- | |- | ||
! LQTS4 | ! LQTS4 | ||
| 4q25 | | 4q25-q27 | ||
| ANK2 | | ANK2 | ||
| Ankyrin B | | Ankyrin B | ||
| | | I''Na,K'' | ||
| <1% | | <1% | ||
| | | | ||
| AD | | AD | ||
| | | | ||
| Line 159: | Line 163: | ||
|- | |- | ||
! LQTS5 | ! LQTS5 | ||
| 21q22 | | 21q22.1 | ||
| KCNE1 | | KCNE1 | ||
| minK | | minK | ||
| Line 172: | Line 176: | ||
|- | |- | ||
! LQTS6 | ! LQTS6 | ||
| 21q22 | | 21q22.1 | ||
| KCNE2 | | KCNE2 | ||
| MiRP1 | | MiRP1 | ||
| Line 184: | Line 188: | ||
| {{OMIM2|603796}} | | {{OMIM2|603796}} | ||
|- | |- | ||
! LQTS7 | ! LQTS7 = ATS1 | ||
| 17q23 | | 17q23 | ||
| KCNJ2 | | KCNJ2 | ||
| Line 197: | Line 201: | ||
| {{OMIM2|600681}} | | {{OMIM2|600681}} | ||
|- | |- | ||
! LQTS8 | ! LQTS8 = TS1 | ||
| | | 12p13.3 | ||
| CACNA1C | | CACNA1C | ||
| | | Ca<sub>v</sub>1.2 | ||
| I''Ca-L'' | | I''Ca-L'' | ||
| <1% | | <1% | ||
| Line 208: | Line 212: | ||
| | | | ||
| Timothy syndrome | | Timothy syndrome | ||
| {{OMIM2| | | {{OMIM2|601005}} | ||
|- | |- | ||
! LQTS9 | ! LQTS9 | ||
| Line 214: | Line 218: | ||
| CAV3 | | CAV3 | ||
| Caveolin 3 | | Caveolin 3 | ||
| | | I''Na'' | ||
| | | | ||
| unknown | | unknown | ||
| Line 226: | Line 230: | ||
| 11q23.3 | | 11q23.3 | ||
| SCN4B | | SCN4B | ||
| | | Na<sub>v</sub>1.5 b4 | ||
| | | | ||
| 1 family | | 1 family | ||
| Line 235: | Line 239: | ||
| | | | ||
| {{OMIM2|608256}} | | {{OMIM2|608256}} | ||
|- | |||
! LQTS11 | |||
| 7q21-q22 | |||
| Akap9 | |||
| AKAP | |||
| I''ks'' | |||
| 1 family | |||
| unknown | |||
| | |||
| | |||
| | |||
| | |||
| {{OMIM2|611820}} | |||
|} | |} | ||
;LQTS: Long QT syndrome | ;LQTS: Long QT syndrome | ||
| Line 240: | Line 257: | ||
;SCD: Sudden Cardiac Death | ;SCD: Sudden Cardiac Death | ||
Long before the genes involved were known, two syndromes | Long before the genes involved were known, two syndromes associated with a prolonged QT interval on the ECG had been described. | ||
* Anton Jervell and Fred Lange-Nielsen from Oslo described in 1957 | * Anton Jervell and Fred Lange-Nielsen from Oslo described in 1957 an autosomaal recessive syndrome that was associated with QT interval prolongation, deafness and sudden death: the now called '''Jervell-Lange-Nielsen syndrome'''. <cite>Lang1957</cite> | ||
* '''Romano-Ward syndrome''' is a long QT syndrome with normal auditory function and autosomal dominant inheritance. | * '''Romano-Ward syndrome''' is a long QT syndrome with normal auditory function and autosomal dominant inheritance. | ||
* In a genotype–phenotype study by Moss et al. that studied type-1 LQTS, it was found that mutations located in the transmembrane portion of the ion channel protein and the degree of ion channel dysfunction caused by the mutations are important independent risk factors influencing the clinical course of this disorder.<cite>moss</cite> | * In a genotype–phenotype study by Moss et al. that studied type-1 LQTS, it was found that mutations located in the transmembrane portion of the ion channel protein and the degree of ion channel dysfunction caused by the mutations are important independent risk factors influencing the clinical course of this disorder.<cite>moss</cite> | ||
Latest revision as of 19:47, 27 August 2020
| Author(s) | J.S.S.G. de Jong, MD | |
| Moderator | P.G. Postema, MD | |
| Supervisor | ||
| some notes about authorship | ||
The Long QT Syndrome (LQTS) is characterized on the ECG by prolongation of the heart rate corrected QT interval. This was first recognized by Dr. Jervell and Dr. Lange-Nielsen in 1957. They described 4 children with a long QT interval which was accompanied by hearing deficits, sudden cardiac death and an autosomal recessive inheritance.Lang1957
The Long QT syndrome may be divided into two distinct forms: congenital Long QT syndrome and acquired Long QT syndrome. These forms may however overlap when QT prolongation due to medication occurs in a patient with congenital Long QT syndrome.
-
 A 12-lead ECG of a patient with acquired long QT syndrome. Notice the QT prolongation. The QTc is about 640ms.
A 12-lead ECG of a patient with acquired long QT syndrome. Notice the QT prolongation. The QTc is about 640ms. -
 A 12 lead ECG of a patient with genetically proven LQTS1
A 12 lead ECG of a patient with genetically proven LQTS1 -
 A 12 lead ECG of a patient with genetically proven LQTS2
A 12 lead ECG of a patient with genetically proven LQTS2 -
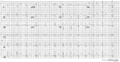 A 12 lead ECG of a patient with genetically proven LQTS3
A 12 lead ECG of a patient with genetically proven LQTS3




Diagnosis
- The diagnosis is by measurement of the heart rate-corrected QT interval on the ECG, which can be calculated with the QTc calculator.
- Sometimes the QT interval can be difficult to assess. Read the guidelines for measurement of difficult QT interval.
- A QTc of > 500ms in patients with Long QT Syndrome is associated with an increased risk for sudden death.Priori
- In patients suspected of LQTS (e.g. family members of known LQTS patients) a QTc > 430ms makes it likely that a LQTS gene defect is present.Hofman
- Because the QTc can change with age, it is best to take the ECG with the longest QTc interval for risk stratification.Goldenberg
TreatmentACC2006
- "Lifestyle modification":
- No competitive sports in all LQTS patients
- No swimming in LQT1 patients
- Avoid nightly noise in LQT2 patients (e.g. no alarm clock)
- Medication: beta-blockers. Beta-blockers even reduce the risk of sudden death in patients in whom a genetic defect has been found, but no QT prolongation is visible on the ECG.
- ICD implantation in combination with beta-blockers in LQTS patients with previous cardiac arrest or syncope or ventricular tachycardia while on beta-blockers.
Acquired LQTS
Acquired LQTS is most often caused by drugs that prolong the QT interval. Combined with risk factors (see table) the risk of Torsade de Pointes increases.
|
|
Congenital LQTS

:
- LQT1 'early onset' broad based T wave
- LQT2 small late T wave
- LQT3 prolonged QT interval with 'late onset' T wave with a normal configuration
In congenital LQTS the ventricular repolarisation is prolonged. The prevalence is about 1:3000-5000.
More than 10 different types of congenital LQTS have been described. However, only LQTS 1-3 are relatively common.ACC2006
| Type | Chromosome | Gene | Protein | Ionchannel | Frequencypriori | SCD incidenceShah2005 | Inheritance | ECG characteristics | Trigger | Eponyme | OMIM™ link |
| LQTS1 | 11p15 | KCNQ1 | KvLQT1 | Iks | ~50% | 0.30%/year | AD, AR | broad base 'early onset' T wave | exercise, especially swimming | JLN1 if homozygous, LQTS1 if heterozygous | 607542 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| LQTS2 | 7q35 | KCNH2 | hERG | Ikr | 30-40% | 0.60%/year | AD | small late T wave | adrenergic triggers, especially nightly noise | JLN2 if homozygous, LQTS2 if heterozygous | 152427 |
| LQTS3 | 3p21 | SCN5A | NA channel | 5-10% | 0.56%/year | AD | 'Late onset' T wave with normal configuration | 600163 | |||
| LQTS4 | 4q25-q27 | ANK2 | Ankyrin B | INa,K | <1% | AD | 106410 | ||||
| LQTS5 | 21q22.1 | KCNE1 | minK | Iks | <1% | unknown | AD/AR | 176261 | |||
| LQTS6 | 21q22.1 | KCNE2 | MiRP1 | Ikr | <1% | unknown | AD | 603796 | |||
| LQTS7 = ATS1 | 17q23 | KCNJ2 | Kir 2.1 | IK1 | <1% | unknown | AD | Anderson-Tawil syndrome | 600681 | ||
| LQTS8 = TS1 | 12p13.3 | CACNA1C | Cav1.2 | ICa-L | <1% | unknown | alternating T waves | Timothy syndrome | 601005 | ||
| LQTS9 | 3p25.3 | CAV3 | Caveolin 3 | INa | unknown | 601253 | |||||
| LQTS10 | 11q23.3 | SCN4B | Nav1.5 b4 | 1 family | unknown | 608256 | |||||
| LQTS11 | 7q21-q22 | Akap9 | AKAP | Iks | 1 family | unknown | 611820 |
- LQTS
- Long QT syndrome
- JLN
- Jervell and Lange-Nielsen syndrome
- SCD
- Sudden Cardiac Death
Long before the genes involved were known, two syndromes associated with a prolonged QT interval on the ECG had been described.
- Anton Jervell and Fred Lange-Nielsen from Oslo described in 1957 an autosomaal recessive syndrome that was associated with QT interval prolongation, deafness and sudden death: the now called Jervell-Lange-Nielsen syndrome. Lang1957
- Romano-Ward syndrome is a long QT syndrome with normal auditory function and autosomal dominant inheritance.
- In a genotype–phenotype study by Moss et al. that studied type-1 LQTS, it was found that mutations located in the transmembrane portion of the ion channel protein and the degree of ion channel dysfunction caused by the mutations are important independent risk factors influencing the clinical course of this disorder.moss
External links
- Torsades.org has a list of QT prolonging drugs
- QTdrugs.org, another list of QT prolonging drugs
- Sudden Arrhythmia Death Syndrome Foundation. LQTS patient group.
- Inherited Arrhythmias Database
Referenties
<biblio>
- Schwartz2001 pmid=11136691
- Shah2005 pmid=16230503
- Lang1957 pmid=13435203
- ACC2006 pmid=16935995
- Goldenberg pmid=16949500
- Roden pmid=14999113
- moss pmid=17470695
- priori pmid=12736279
- Hofman pmid=17090615
- Roden pmid=18184962
</biblio>